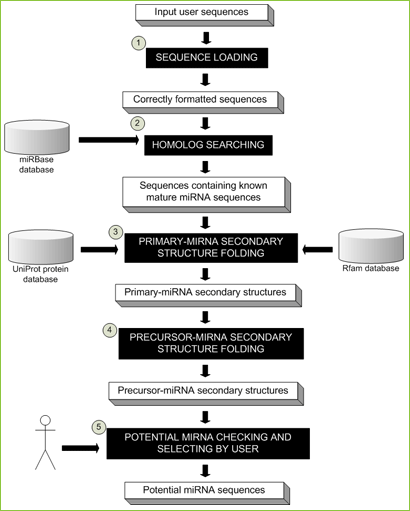
Figure 1 MiRNA identification pipeline
|
MiRNA identification pipeline:
- An input sequence file in FASTA format will be loaded and checked. Sequences which are longer than 3000 bps and/or containing A, T, G, C, U, and N will be excluded from following steps.
- Remaining sequences from the first step will be searched, with user-defined parameters, against known plant mature miRNAs from miRBase [1] for miRNA homologs.
- Sequences containing plant mature miRNAs will be optionally searched against the UniProt [2] and Rfam [3] databases to remove protein-coding sequences and other types of non-coding RNAs, respectively. This step is performed by BLASTX and BLASTN from BLAST package [4] with user defined expected values and databases. Then, remaining sequences will be predicted for the secondary structures of primary miRNAs (pri-miRNAs) by UNAFold [5] with user defined parameters.
- Precursor miRNAs (pre-miRNAs), short stem-loop structures, will be cleaved from pri-miRNAs. Users could manually specify start and stop positions for pre-miRNA cleavage or select an automatic option, which pre-miRNAs with two nucleotides at 3' overhang will be cleaved automatically.
- Potential miRNAs will be decided by users based on previous steps' results including (1) number of mismatches between known and predicted mature miRNA sequences, (2) number of mismatches between predicted mature miRNA and its miRNA* sequences, (3) number and size of bulges within miRNA-miRNA* duplex, and (4) minimal free energies (MFEs) and minimal folding free energy index (MFEI) [6].
|
|
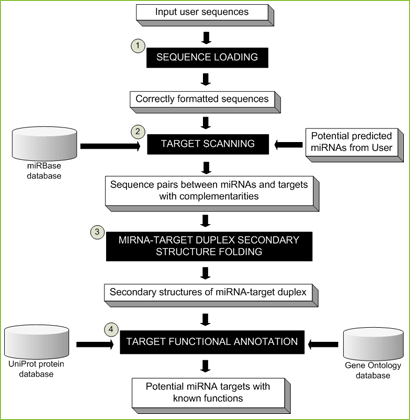
Figure 2 MiRNA target identification pipeline
|
MiRNA target identification pipeplie:
- An input sequence file in FASTA format will be loaded and checked, similar to the seqeuence loading step in miRNA identification.
- Remaining sequences from the first step will be scanned for the complementary sites of either known plant mature miRNAs from miRBase and/or potential miRNAs from miRNA identification.
- MiRNA-target pairs with complementary sites will be predicted for their duplex secondary structures by UNAFold with user-defined parameters.
- Sequences containing miRNA complementary sites will be assigned functions by homolog search against the UniProt protein database and mapped functional category by Gene Ontology. Potential miRNA targets will be decided by users based on previous steps' results including (1) number of mismatches between miRNA and its potential target at the complementary site, (2) number of G:U pairs, (3) mismatched positions, and (4) minimal free energies (MFEs).
References
- Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ: miRBase: microRNA sequences, targets and gene nomenclature. Nucl Acids Res 2006, 34(suppl_1):D140-144.
- The UniProt Consortium: The Universal Protein Resource (UniProt). Nucl Acids Res 2008, 36(suppl_1):D190-195.
- Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A: Rfam: annotating non-coding RNAs in complete genomes. Nucl Acids Res 2005, 33(suppl_1):D121-124
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. Journal of Molecular Biology 1990, 215:403-4101
- Markham NR, Zuker M: UNAFold: software for nucleic acid folding and hybriziation . In: Bioinformatics, Structure, Functions and Applications. Edited by Keith JM, Totowa NJ, vol. II: Humana Press; 2008: 3-30.
- Zhang B, Pan X, Cox S, Cobb G, Anderson T: Evidence that miRNAs are different from other RNAs. Cellular and Molecular Life Sciences (CMLS) 2006, 63(2):246-254.
|
|
 |
|
|
|
